Congenital defects of the nervous system: Difference between revisions


m (citation corrected by the editor) |
m (Checked by the editor, grammar has NOT been corrected) |
||
| (2 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
__noTOC__ | __noTOC__ | ||
==[[Arachnoid cysts|Arachnoid cyst]]== | ==[[Arachnoid cysts|Arachnoid cyst]]== | ||
{{Edit article|Arachnoid cysts}}{{:Arachnoid cysts}} | |||
| | |||
}} | |||
Arachnoid cysts | |||
== '''[[Chiari malformation]]''' == | == '''[[Chiari malformation]]''' == | ||
{{Edit article|Chiari malformation}}{{:Chiari malformation}} | |||
== [[Craniostenosis]] == | == [[Craniostenosis]] == | ||
{{Edit article|Craniostenosis}}{{:Craniostenosis}} | |||
{{ | |||
==[[Meningocele]]== | ==[[Meningocele]]== | ||
{{Edit article|Meningocele}}{{:Meningocele}} | |||
== [[Neurofibromatosis]]== | == [[Neurofibromatosis]]== | ||
{{Edit article|Neurofibromatosis}}{{:Neurofibromatosis}} | |||
{{ | |||
==[[Dandy-Walker syndrome]]== | ==[[Dandy-Walker syndrome]]== | ||
Dandy-Walker syndrome | {{Edit article|Dandy-Walker syndrome}}{{:Dandy-Walker syndrome}} | ||
}} | |||
}} | |||
==[[Cerebral palsy in children|Cerebral palsy in children]]== | |||
==[[ | |||
__toc__ | __toc__ | ||
Cerebral palsy is a '''permanent''', '''non-progressive movement disorder''' accompanied by '''abnormal muscle tension and abnormal posture'''. | |||
It is a '''non'''-'''contagious''', '''non'''-'''hereditary''' '''disease''' that arises on the basis of one-time damage to brain tissue (most often hypoxia). | |||
It is '''characterized''' by a disorder in '''the development of the motor areas''' of the brain '''or''' their other '''damage at an early stage of development'''. Cerebral palsy is '''the''' '''result''' of prenatal, postnatal, or early postnatal damage to the developing brain. A movement disorder is often accompanied by epilepsy, disorders of sensitivity, senses (e.g. visual impairment) and perception, learning disorders, cognition, communication, behavior or mental retardation. Cerebral palsy is a neurodevelopmental disorder whose manifestations usually change during development. | |||
Cerebral palsy belongs to the group of developmental diseases because it arises on the basis of a wide spectrum of abnormalities of the developing CNS. Different etiologies acting on different developmental stages can lead to the same clinical picture, and conversely, a similar etiology can cause different consequences. | Cerebral palsy belongs to the group of '''developmental diseases''' because it arises on the basis of a wide spectrum of abnormalities of the developing CNS. Different etiologies acting on different developmental stages can lead to the same clinical picture, and conversely, a similar etiology can cause different consequences. | ||
Cerebral palsy treatment is complex, multidisciplinary and long-term. Its aim is not to cure or achieve a normal state, but to increase functionality, improve abilities and maintain health in terms of locomotion, cognitive development, social integration and independence. The success of therapy depends on its timeliness and intensity | Cerebral palsy '''treatment''' is '''complex''', '''multidisciplinary''' and '''long'''-'''term'''. Its aim is not to cure or achieve a normal state, but to increase functionality, improve abilities and maintain health in terms of locomotion, cognitive development, social integration and independence. The success of therapy depends on its timeliness and intensity | ||
'''Not to be confused with poliomyelitis anterior acute, the so-called poliomyelitis!''' | |||
== Classification == | == Classification == | ||
* according to the anatomical topography of the disability: | * according to the anatomical topography of the disability: | ||
| Line 263: | Line 44: | ||
==Etiology== | ==Etiology== | ||
[[File:1426_Corticospinal_Pathway.jpg|thumb|Kortikospinální dráhy.]]Movement disorder in cerebral palsy is caused by involvement of supraspinal movement centers, corticospinal tracts, segmental spinal circuits and the musculoskeletal system.<ref name="Kraus"/> | [[File:1426_Corticospinal_Pathway.jpg|thumb|Kortikospinální dráhy.]]'''Movement''' '''disorder''' in cerebral palsy is '''caused''' by '''involvement of supraspinal movement''' centers, corticospinal tracts, segmental spinal circuits and the musculoskeletal system.<ref name="Kraus"/> | ||
It can arise during one of the four following periods: | It can arise during one of the four following periods: | ||
*During pregnancy (prenatal etiology) – intrauterine infection, drugs or placental dysfunction; | *'''During''' '''pregnancy''' (prenatal etiology) – intrauterine infection, drugs or placental dysfunction; | ||
*During childbirth (perinatal etiology) - mainly in multiple pregnancies, hypoxia, hemorrhage, hypoglycemia, meningitis; | *'''During''' '''childbirth''' (perinatal etiology) - mainly in multiple pregnancies, hypoxia, hemorrhage, hypoglycemia, meningitis; | ||
*In the first months of a child's life (postnatal etiology) – trauma, encephalopathy, encephalitis; | *'''In the first months of a child's life''' (postnatal etiology) – trauma, encephalopathy, encephalitis; | ||
*Or very low birth weight. | *'''Or very low birth weight'''. | ||
In the first and second trimester, disorders of CNS development occur. At the beginning of the third trimester, it is periventricular leukomalacia (PVL) and intraventricular hemorrhage (IVH). Towards the end of the third trimester, cortical, subcortical and deep gray matter lesions appear..<ref name="Kraus"/> | In the first and second trimester, disorders of CNS development occur. At the beginning of the third trimester, it is periventricular leukomalacia (PVL) and intraventricular hemorrhage (IVH). Towards the end of the third trimester, cortical, subcortical and deep gray matter lesions appear..<ref name="Kraus"/> | ||
*Bilateral spastic and dyskinetic forms – hypoxic-ischemic brain lesion. | *'''Bilateral''' '''spastic and dyskinetic forms''' – hypoxic-ischemic brain lesion. | ||
*Diparesis – periventricular leukomalacia in premature infants. | *'''Diparesis''' – periventricular leukomalacia in premature infants. | ||
*Spastic hemiparesis of mature newborns - middle cerebral artery infarction, periventricular gliosis. | *'''Spastic hemiparesis of mature newborns''' - middle cerebral artery infarction, periventricular gliosis. | ||
*Spastic hemiparesis of premature newborns - periventricular porencephaly due to intraventricular hemorrhage. | *'''Spastic hemiparesis of premature newborns''' - periventricular porencephaly due to intraventricular hemorrhage. | ||
*Dyskinetic form – hypoxic-ischemic damage in the area of the thalamus and basal ganglia (children after the 32nd week of pregnancy). | *'''Dyskinetic''' '''form''' – hypoxic-ischemic damage in the area of the thalamus and basal ganglia (children after the 32nd week of pregnancy). | ||
*Athetoid form - neonatal hyperbilirubinemia with nuclear icterus (very rare). | *'''Athetoid''' '''form''' - neonatal hyperbilirubinemia with nuclear icterus (very rare). | ||
*Atactic form - mostly unknown cause, possibly structural disorder of the cerebellum.<ref name="muntau" /> | *'''Atactic''' form - mostly unknown cause, possibly structural disorder of the cerebellum.<ref name="muntau" /> | ||
==Clinical symptoms== | ==Clinical symptoms== | ||
| Line 289: | Line 69: | ||
* level of disability: GMFCS scale I – V (Gross Motor Function Classification System) – a functional test for assessing the degree and subsequent changes in gross motor skills using standard free movements (free range of motion, walking and sitting); MACS (The Manual Ability Classification System) – test of manual abilities; | * level of disability: GMFCS scale I – V (Gross Motor Function Classification System) – a functional test for assessing the degree and subsequent changes in gross motor skills using standard free movements (free range of motion, walking and sitting); MACS (The Manual Ability Classification System) – test of manual abilities; | ||
* comorbidities: epilepsy, mental retardation, sensory disorders.<ref name="Kraus">{{Cite| type = article| surname1 = Kraus| name1 = Josef| article = Dětská mozková obrna| journal = Neurol. praxi| url = https://www.neurologiepropraxi.cz/pdfs/neu/2011/04/02.pdf| year = 2011| the_year = 12| edition = 4| pages = 222–224| issn = -}}</ref> | * comorbidities: epilepsy, mental retardation, sensory disorders.<ref name="Kraus">{{Cite| type = article| surname1 = Kraus| name1 = Josef| article = Dětská mozková obrna| journal = Neurol. praxi| url = https://www.neurologiepropraxi.cz/pdfs/neu/2011/04/02.pdf| year = 2011| the_year = 12| edition = 4| pages = 222–224| issn = -}}</ref> | ||
* Spasticity: abnormally increased muscle tone, increased muscle reflexes, positive pyramidal phenomena, abnormal movements and postures (pes equinus, internal rotation and adduction of the hips, pronation and flexion of the forearm), the formation of contractures. | * Spasticity: abnormally increased muscle tone, increased muscle reflexes, positive pyramidal phenomena, abnormal movements and postures (pes equinus, internal rotation and adduction of the hips, pronation and flexion of the forearm), the formation of contractures. | ||
| Line 298: | Line 77: | ||
===Spastic forms=== | ===Spastic forms=== | ||
====Diparetic form (sometimes referred to as parapetic)==== | ====Diparetic form (sometimes referred to as parapetic)==== | ||
*Most common (1/3 affected). | *Most common (1/3 affected). | ||
*Symmetrical involvement of both lower limbs (weaker, less developed) | *Symmetrical involvement of both lower limbs (weaker, less developed) | ||
| Line 306: | Line 84: | ||
====Hemiparetic form==== | ====Hemiparetic form==== | ||
*The second most common | *The second most common | ||
*Affected limbs weaker and usually shorter compared to the other side. | *Affected limbs weaker and usually shorter compared to the other side. | ||
| Line 314: | Line 91: | ||
====Quadruparetic form==== | ====Quadruparetic form==== | ||
*A more severe form of the diparetic form. | *A more severe form of the diparetic form. | ||
*All 4 limbs affected by polio. | *All 4 limbs affected by polio. | ||
| Line 320: | Line 96: | ||
===Dyskinetic-atactic forms=== | ===Dyskinetic-atactic forms=== | ||
*About 10-15% cases | *About 10-15% cases | ||
*Epilepsy is rare in these forms. | *Epilepsy is rare in these forms. | ||
| Line 329: | Line 104: | ||
==Diagnostics== | ==Diagnostics== | ||
*Cerebral palsy is a '''clinical''' diagnosis; '''it is definitively determined only after the 3rd year of age''' (with immaturity of the CNS, the manifestations are non-specific). | *Cerebral palsy is a '''clinical''' diagnosis; '''it is definitively determined only after the 3rd year of age''' (with immaturity of the CNS, the manifestations are non-specific). | ||
*Examination to clarify the etiology: | *Examination to clarify the etiology: | ||
| Line 345: | Line 119: | ||
===A treatment plan may include:=== | ===A treatment plan may include:=== | ||
*Rehabilitation – improving control of locomotion and postural posture using stimulation and facilitation techniques (e.g. the Vojt method), first by experts, then by instructed parents. | *Rehabilitation – improving control of locomotion and postural posture using stimulation and facilitation techniques (e.g. the Vojt method), first by experts, then by instructed parents. | ||
*[[ | *[[Antiepileptics|Antiepileptic drugs.]] | ||
*Myorelaxans in spastic forms. | *Myorelaxans in spastic forms. | ||
*Splints for muscle imbalances. | *Splints for muscle imbalances. | ||
| Line 357: | Line 131: | ||
== Links == | == Links == | ||
=== Related Articles === | === Related Articles === | ||
* [[Congenital developmental defects]] | * [[Congenital developmental defects of the newborn requiring an urgent solution|Congenital developmental defects]] | ||
=== Source === | === Source === | ||
* {{Cite|type = web|surname1 = Beneš|name1 = Jiří|source_name = Studijní materiály|year = 2007|cited = 2010|url = http://jirben.wz.cz}} | * {{Cite|type = web|surname1 = Beneš|name1 = Jiří|source_name = Studijní materiály|year = 2007|cited = 2010|url = http://jirben.wz.cz}} | ||
Latest revision as of 20:38, 20 December 2022
Arachnoid cyst
An arachnoid cyst is a well-defined cystic, congenital collection of cerebrospinal fluid (it arises on the basis of doubling of the arachnoid layer). It is often an accidental finding on a CT scan or MRI scan. It is important for a differentia diagnosis of (tumors).[1] [2]
Symptoms and auxiliary tests
In the CT image (resp MRI, possibly with the intrathecal application of contrast) arachnoid cyst has a density, resp. cerebrospinal fluid intensity (hypodense). Most common locations:
- in the middle pit
- on convexity
- in the posterior cranial pit (cisterna magna)
- in the cerebellopontine angle
- at the chiasm level
- in the chamber
Expansive behaviour depends on its communication with the surrounding cerebrospinal fluid spaces. The problem occurs if the communication has a valve character (intracranial hypertension syndrome) occurs.
Arachnoid cysts can be asymptomatic, otherwise, they often cause symptoms in early childhood (increased intracranial pressure, epileptic seizures, gradually developing focal neurological symptoms, sudden worsening of the condition during cyst bleeding.[1][2]
- CT of arachnoid cysts of the temporal lobe

Sagittal plane

Frontal plane
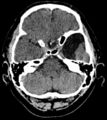
Transverse plane



Treatment
An incidental finding and an asymptomatic cyst are not treated or followed up. For expansive behaviour and clinical manifestations, surgery is chosen (shunt surrounding cerebrospinal fluid, ie microsurgical / endoscopic fenestration of the cyst into the normal cerebrospinal fluid). A shunt formed between the arachnoid cyst (most often in the case of the sulcus lateralis cyst) and the peritoneal cavity, the so-called cystoperitoneal shunt, is also possible. Marsupialization (removal of the cyst with the capsule) is rarely performed.[1][2]
Chiari malformation

Chiari malformation (Arnold-Chiari malformation) is a congenital CNS anomaly. It is a dystopia of the cerebellum and medulla oblongata into the spinal canal, which is clinically manifested by hydrocephalus. RWe distinguish four types of rhombencephalon abnormalities (cerebellum, pons, oblongata):
- Type 1 - herniation of the cerebellar tonsil under the foramen magnum, IV. the chamber is stored normally
- Type 2 - usually the co-presence of myelomeningocele
- Type 3 - severe dislocation of structures in the posterior pit, often associated with suboccipital encephalomeningocele; usually incompatible with life
- Type 4 - cerebellar hypoplasia without herniation.

Clinical picture
Clinically, the defect is manifested mainly by headache, weakened grip and spasticity of DK.
Diagnosis
Native X-ray, MRI.
Therapy
The main problem is hydrocephalus – decompression of the craniospinal junction, short-circuit drainage operations.
Craniostenosis

Craniostenosis is caused by premature adhesion of sutures. The earlier it occurs, the more severe its effects. The shape of the head depends on the order in which the sutures close - as a suture grows, the skull stops growing perpendicular to the suture.
Normal development of the skull and sutures
- at birth, the skull has one lamina, and the diploe is formed at 4 years of age
- large fontanelle is 4x2.5 cm at birth, closes at 1.5-2.5 years
- the small one closes at 2-3 months
- the growth of the sutures is prevented by the proliferation of fibroblasts along the suture line
- CI - cephalic index - percentage ratio of head width to length - 60-70% in children, 70-90% in adults
- lengthening of the head decreases CI
Dividing
- primary - due to a developmental disorder of unclear etiology;
- secondary - in microcephaly and after drainage hydrocephalus.
Diagnosis
- clinically - palpation of fontanelles, X-ray, CT
- at high pressure, gyral impressions are seen on the skull bones - the so-called skull of wrought silver
- scintigraphy - open suture receives Tc, closed not
Types
- scaphocephaly - sagittal suture fusion, the head is narrow, resembles a ship's keel
- brachycephaly - affects the coronal suture - flat forehead, greater distance between orbits, etc.
Therapy
- many surgical methods, indications are intracranial hypertension or cosmetic
- early surgery can ensure normal psychomotor development
Meningocele

Meningocele (spina bifida cystica) is a split of the vertebral arch, from which emerges a sac made up of soft spinal cords (the dura mater usually ends at the neck of the sac). The content is cerebrospinal fluid, 1/3 of children are neurologically affected.
'Menigomyelocele (spina bifida aperta) is a variant of meningocele that occurs a little more often and is less favorable. In addition to the packages, there is also spinal cord in the hernial sac.
Therapy
On admission, place the child on the stomach and exclude pressure on the meningomyelocele. In case of perforation, apply ATB and cover the bearing with a mule. We operate within 24 hours. In complete motor paraplegia and sphincter lesions, this cannot be improved by any surgery, but surgery is in order to prevent infection.
Done by: Eisa Jbara
Neurofibromatosis
Neurofibromatosis is a relatively common AD hereditary disease (1:2,500–4,000 newborns) based on cells derived from the neural bar. It is manifested by abnormal growth of CNS and PNS support cells (Schwann's bb. etc.) with a pronounced predisposition to the formation of benign and malignant tumors[3].
The disease belongs to hereditary tumor syndromes, it occurs in two forms. Familial forms of these syndromes arise as a result of a congenital mutation of tumor-suppressor genes. A certain percentage of these syndromes arise as a result of new mutations[4]. Molecular-genetic analysis is available for definitive confirmation of this diagnosis.
Forms
There are two basic forms of this disease, which differ both in the cause (mutation of different genes) and in the consequences (different clinical picture).
Neurofibromatosis – Type 1
Neurofibromatosis – type 1 (NF-1, also called morbus von Recklinghausen or peripheral type of neurofibromatosis) is conditioned by a mutation of the NF1 gene on chromosome 17 (17q11.2)[5]. It is a tumor-suppressor gene whose product (neurofibromin) is part of the intracellular signaling cascade associated with THE RAS-kinase.
![]() It is necessary to distinguish morbus Recklinghausen, which is synonymous with primary hyperparathyroidism.
It is necessary to distinguish morbus Recklinghausen, which is synonymous with primary hyperparathyroidism.
Clinical manifestations of this form include:
- So-called "café-au-lait spots" (spots of the color of "white coffee", in 90% appear up to 5 years of age).[4]
- Neurofibromas (multiple tumorous nodules; cutaneous, subcutaneous and plexiform; mainly in the axillae and groin).
- Lisch nodules (hamartomy iris).[4][5]
- Increased risk of developing various cancers: CNS gliomas (optics gliomas), neurofibrosarcoma, rhabdomyosarcoma, pheochromocytoma, leukemia, etc.[4]
- Involvement of the musculoskeletal system (subperiosteal neurofibromas – causing hypertrophy of the bone, its thinning and pathological fractures, scoliosis, congenital dysplasia of the tibia)[3].
- Intellectual impairment, epilepsy or stenosis of the renal artery.[4][5]
Neurofibromatosis – Type 2
Neurofibromatosis – type 2 (NF-2, also called MISME syndrome or central type neurofibromatosis) is conditioned by mutation of the NF2 gene on chromosome 22 (22q12.2)[5]. It is also a tumor-suppressor gene whose product (neurofibromin 2, also called merlin or schwannomine) affects intercellular contacts. Central neurofibromatosis is generally rarer than the peripheral type, but overall it is associated with higher morbidity and mortality of affected individuals.[4] About half of NF-2 cases are caused by a new mutation.[5]
Clinical manifestations of this form include:
- CNS tumors: meningeomas, astrocytomas, ependymomas, spinal root schwanoms, retinal hamartoms (MISME syndrome = Multiple Inherited Schwannomas, Meningiomas, and Ependymomas).
- Bilateral vestibular schwannoma is particularly typical.
- Also in this form we find "café-au-lait" spots, but not Lisch's nodules.[4]
Therapy
- Causal therapy does not exist.
- Dispensary of patients with a proven diagnosis of neurofibromatosis is suitable.
- Surgical interventions indicated in case of nerve oppression / obstruction in the GIT form, possibly from a cosmetic point of view.[3]
- Neurosurgical interventions in CNS involvement; possible use of stereotactic neurosurgery (Leksell's gamma knife).
Dandy-Walker syndrome
Dandy-Walker syndrome is a rare disease, affecting the development of the brain. It manifests with a triad of symptoms
- complete or partial agenesis of vermis cerebellum
- cystic dilatation of the fourth ventricle of brain
- enlargement of the posterior cranial fossa[6]
Enlargement of the posterior cranial fossa may be associated with membranous atrasia of the apertures of the fourth ventricle, leading to pathological accumulation of cerebrospinal fluid in the ventricles.
These abnormalities most often result in problems with movement, coordination but also with intellect. Psychiatric illness may also occur[7].
Affected individulas may survive up to their second decade of life.
Malformations
In most cases, individuals with Dandy-Walker syndrome develop symptoms of abnormal brain development during the first year of life. Clinically, it is manifested in most children by the accumulation of cerebrospinal fluid in the brain - hydrocephalus, which can cause macrocephaly.
Mental disability is often present[8], although some individuals also have a normal intellect. Children often have delayed development, especially in the field of motor skills (crawling, walking, coordination of movements). Muscle stiffness and paralysis - spastic paraplegia - may occur. Older children experience symptoms of increased intracranial pressure such as irritability, vomiting, convulsions and nystagmys. Less frequently other cerebral malformations are present such as agenesis of the corpus callosum connecting the right and left hemispheres, occipital encephalocele or abnormal clefts in the brain - schizencephaly and gyrification disorders. These brain defects are associated with more or less severe signs and symptoms. Dandy-Walker syndrome can also include heart defects, malformations of the urogenital tract, limbs and face. A milder form of the disease is the Dandy – Walker variant, which includes hypoplasia of the cerebellar vermis without agenesis, mild or no enlargement of the posterior fossa and fourth ventricle, without hydrocephalus.
Genetics
Mutations in certain genes occur in individuals affected by Dandy-Walker syndrome, but these mutations account for only a small number of all known cases. Dandy-Walker syndrome is also associated with chromosomal abnormalities on most chromosomes.'' It is most common in people with trisomy 18, but can also occur with trisomy 13, 21 or 9. Dandy-Walker syndrome has also been reported in fetuses with triploidy, a fatal condition in which individuals have an extra complete set of chromosomes in each cell. Dandy-Walker syndrome can also be caused by mutations of specific genes (FOXC1 contains instructions for the preparation of a protein that regulates the activity of other genes, ZIC1 and ZIC4 act as transcriptional activators and participate in CNS organogenesis). Brain malformations associated with Dandy-Walker syndrome can occur also in isolation, and may not be associated with a genetic disease, with the cause being often unknown.
Environmental influence
Dandy-Walker syndrome can be caused by environmental factors that affect the fetus before birth. For example exposure of the fetus to rubella, toxoplasmosis or due to substances that causes damage to the fetus, (teratogens). A more frequent occurrence of fetuses with Dandy-Walker syndrome also occurs in the fetuses of mothers who suffer from diabetes.
Diagnosis and incidences
Prenatal diagnosis is possible by ultrasound examination and performing amniocentesis to examine the karyotype of the fetus. It is estimated that Dandy-Walker syndrome occurs in 1:25000 to 1:30000 newborns. The occurrence of cases of Dandy-Walker syndrome is rare and does not have a clear pattern of inheritance. Only immediate relatives of people with Dandy-Walker syndrome have an increased risk of developing the disease compared to the general population.
Therapy
Therapy consists in removing the obstruction, and hydrocephalus is treated by introducing a shunt.
Cerebral palsy in children
Cerebral palsy is a permanent, non-progressive movement disorder accompanied by abnormal muscle tension and abnormal posture.
It is a non-contagious, non-hereditary disease that arises on the basis of one-time damage to brain tissue (most often hypoxia).
It is characterized by a disorder in the development of the motor areas of the brain or their other damage at an early stage of development. Cerebral palsy is the result of prenatal, postnatal, or early postnatal damage to the developing brain. A movement disorder is often accompanied by epilepsy, disorders of sensitivity, senses (e.g. visual impairment) and perception, learning disorders, cognition, communication, behavior or mental retardation. Cerebral palsy is a neurodevelopmental disorder whose manifestations usually change during development.
Cerebral palsy belongs to the group of developmental diseases because it arises on the basis of a wide spectrum of abnormalities of the developing CNS. Different etiologies acting on different developmental stages can lead to the same clinical picture, and conversely, a similar etiology can cause different consequences.
Cerebral palsy treatment is complex, multidisciplinary and long-term. Its aim is not to cure or achieve a normal state, but to increase functionality, improve abilities and maintain health in terms of locomotion, cognitive development, social integration and independence. The success of therapy depends on its timeliness and intensity
Not to be confused with poliomyelitis anterior acute, the so-called poliomyelitis!
Classification
- according to the anatomical topography of the disability:
- mono-, hemi-, di-, quadruparetic form;
- according to the pathophysiological type of movement disorder:
- spastic forms and non-spastic forms: with dyskinesias, dystonias, hypotonia, ataxia.[9]
Epidemiology
The incidence of Cerebral palsy is about 2:1000 live births. The risk increases inversely with the gestational age at delivery.[10]
Etiology

Movement disorder in cerebral palsy is caused by involvement of supraspinal movement centers, corticospinal tracts, segmental spinal circuits and the musculoskeletal system.[9]
It can arise during one of the four following periods:
- During pregnancy (prenatal etiology) – intrauterine infection, drugs or placental dysfunction;
- During childbirth (perinatal etiology) - mainly in multiple pregnancies, hypoxia, hemorrhage, hypoglycemia, meningitis;
- In the first months of a child's life (postnatal etiology) – trauma, encephalopathy, encephalitis;
- Or very low birth weight.
In the first and second trimester, disorders of CNS development occur. At the beginning of the third trimester, it is periventricular leukomalacia (PVL) and intraventricular hemorrhage (IVH). Towards the end of the third trimester, cortical, subcortical and deep gray matter lesions appear..[9]
- Bilateral spastic and dyskinetic forms – hypoxic-ischemic brain lesion.
- Diparesis – periventricular leukomalacia in premature infants.
- Spastic hemiparesis of mature newborns - middle cerebral artery infarction, periventricular gliosis.
- Spastic hemiparesis of premature newborns - periventricular porencephaly due to intraventricular hemorrhage.
- Dyskinetic form – hypoxic-ischemic damage in the area of the thalamus and basal ganglia (children after the 32nd week of pregnancy).
- Athetoid form - neonatal hyperbilirubinemia with nuclear icterus (very rare).
- Atactic form - mostly unknown cause, possibly structural disorder of the cerebellum.[10]
Clinical symptoms

- form: spastic, dyskinetic or ataxic;
- distribution: bilateral – diparesis, quadruparesis, unilateral – hemiparesis
- level of disability: GMFCS scale I – V (Gross Motor Function Classification System) – a functional test for assessing the degree and subsequent changes in gross motor skills using standard free movements (free range of motion, walking and sitting); MACS (The Manual Ability Classification System) – test of manual abilities;
- comorbidities: epilepsy, mental retardation, sensory disorders.[9]
- Spasticity: abnormally increased muscle tone, increased muscle reflexes, positive pyramidal phenomena, abnormal movements and postures (pes equinus, internal rotation and adduction of the hips, pronation and flexion of the forearm), the formation of contractures.
- Dystonia: abnormal sustained muscle contractures that lead to abnormal dystonic posture and abnormal movements (flexion, wrist pronation with fingers extended, or trunk torsion).
- Athetosis: generalized, uncoordinated, exaggerated, involuntary hyperkinetic movements, normal or reduced muscle tone.
- Ataxia: dysmetria or intentional tremor of the upper limbs, ataxia of walking and standing - involvement of the lower limbs and trunk (standing and walking with a wide base, swaying gait).[10]
Spastic forms
Diparetic form (sometimes referred to as parapetic)
- Most common (1/3 affected).
- Symmetrical involvement of both lower limbs (weaker, less developed)
- Striking disproportion between the growth of the trunk and the lower limbs.
- Muscle hypertonia, muscle shunts - faulty posture of the lower limbs and pelvis.
- There may be mild to moderate mental retardation
Hemiparetic form
- The second most common
- Affected limbs weaker and usually shorter compared to the other side.
- The upper limb is almost always more affected.
- A typical impairment is characterized by the arm being drawn to the torso, bent to full flexion at the elbow, the forearm turned dorsally upwards, the hand bent towards the palm and averted towards the little finger side.
- Epilepsy is common, or there may also be sensory deficits and mental retardation.
Quadruparetic form
- A more severe form of the diparetic form.
- All 4 limbs affected by polio.
- Epilepsy and mental retardation or learning disabilities are also present.
Dyskinetic-atactic forms
- About 10-15% cases
- Epilepsy is rare in these forms.
- If choreoathetosis predominates, intelligence is usually normal, but verbal expression is made difficult by severe dysarthria.
- If dystonia or ataxia predominates, mental retardation is more often present.
The disease is often a combination of several forms of cerebral palsy.
Diagnostics
- Cerebral palsy is a clinical diagnosis; it is definitively determined only after the 3rd year of age (with immaturity of the CNS, the manifestations are non-specific).
- Examination to clarify the etiology:
- US of the brain, MRI of the brain;
- coagulation (protein C and S, homocysteine, APC resistance);
- EEG; vision and hearing tests;
- genetic examination - for lissencephaly[10]
Differential diagnosis
A stable clinical finding without progression of disability is mainly indicative of cerebral palsy. An imaging examination (CT, MRI) is important to determine the extent. The progression of the disability is more likely to indicate a neurodegenerative or neurometabolic disability. A slow-growing tumor of the CNS.
Therapy
Cerebral palsy is a disease that cannot be cured. However, it is possible to improve the conditions and life possibilities of the child with the help of treatment, which usually positively affects the quality of his life. Medical advances in recent years, and especially advances in the treatment of people with cerebral palsy, have led to the fact that today many of those who have been treated in time and correctly can lead an almost normal life. Even today, there is still no standard treatment that is sufficiently effective for all patients. A doctor who treats a patient with cerebral palsy is more or less dependent on a number of specialized experts, with whose help he first correctly identifies and determines individual disorders and then adapts the entire therapeutic program to them. Several clinical trials are also currently underway on the application of stem cells from umbilical cord blood.
A treatment plan may include:
- Rehabilitation – improving control of locomotion and postural posture using stimulation and facilitation techniques (e.g. the Vojt method), first by experts, then by instructed parents.
- Antiepileptic drugs.
- Myorelaxans in spastic forms.
- Splints for muscle imbalances.
- Operative treatment – orthopedic corrections (between 6 and 10 years), for example, in the diparetic form, selective dorsal rhizotomy (partial transection of the posterior spinal roots at the level of L2).
- Prosthetic aids.
- Application of botulinum toxin.
- Special care and education from preschool age.
Links
Related Articles
Source
- BENEŠ, Jiří. Studijní materiály [online]. ©2007. [cit. 2010]. <http://jirben.wz.cz>.
References
- ↑ Jump up to: a b c SEIDL, Zdeněk – OBENBERGER, Jiří. Neurologie pro studium i praxi. 1. edition. Praha : Grada Publishing, 2004. ISBN 80-247-0623-7.
- ↑ Jump up to: a b c SAMEŠ, M, et al. Neurochirurgie. 1. edition. Praha : Jessenius Maxdorf, 2005. ISBN 80-7345-072-0.
- ↑ Jump up to: a b c DUNGL, P., et al. Ortopedie. 1. vydání. Praha : Grada Publishing, 2005. ISBN 80-247-0550-8.
- ↑ Jump up to: a b c d e f g KLEIBL, Zdeněk a Jan NOVOTNÝ. Hereditární nádorové syndromy. 1. vydání. Praha : Triton, 2003. 31 s. ISBN 80-7254-357-1.
- ↑ Jump up to: a b c d e FIRTH, Helen V., Jane A. HURST a Judith G. HALL. Oxford desk reference: clinical genetics. 1. vydání. Oxford : Oxford University Press, 2005. 708 s. ISBN 9780192628961.
- ↑ MARCDANTE, Karen J. Nelson essentials of pediatrics. 7. edition. 2015. 0 pp. ISBN 978-1-4557-5980-4.
- ↑ CAN, Serdar Suleyman – KARAKAŞ UĞURLU, Görkem – CAKMAK, Selcen. Dandy walker variant and bipolar I disorder with graphomania. Psychiatry Investig [online]. 2014, vol. 11, no. 3, p. 336-9, Available from <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4124195/?tool=pubmed>. ISSN 1738-3684. DOI: 10.4306/pi.2014.11.3.336.
- ↑ PANDURANGI, Swapna – PANDURANGI, Aditya – MATKAR, Abhay. Psychiatric manifestations associated with mega cisterna magna. J Neuropsychiatry Clin Neurosci [online]. 2014, vol. 26, no. 2, p. 169-71, Available from <https://www.ncbi.nlm.nih.gov/pubmed/24763763>. ISSN 0895-0172 (print), 1545-7222.
- ↑ Jump up to: a b c d KRAUS, Josef. Dětská mozková obrna. Neurol. praxi [online]. 2011, y. 12, p. 222–224, Available from <https://www.neurologiepropraxi.cz/pdfs/neu/2011/04/02.pdf>.
- ↑ Jump up to: a b c d MUNTAU, Ania Carolina. Pediatrie. 4. edition. Grada, 2009. pp. 521-523. ISBN 978-80-247-2525-3.
