Polycystic kidney disease


Polycystic kidney disease (PKD) is a disease characterized by the presence of cysts in the renal cortex and / or parenchyma. The presence of the cysts leads to the loss of functional renal parenchyma and may result in renal insufficiency. Based on the mode of inheritance, we can divide the disease into autosomal dominant and autosomal recessive PKD.
Autosomal dominant polycystic kidney disease[edit | edit source]
Autosomal dominant polycystic kidney disease (ADPKD), or adult PKD, is the most common congenital renal disease (incidence 1:1000). There are numerous cortical and medullary cysts that grow and lead to a reduction of the functional parenchyma.
This is most often a mutation in the PKD1 gene on chromosome 16 (90%). The pathological structural glycoprotein polycystin is formed, which is a building block of the basement membrane of the renal tubules. During the course of life, these incomplete tubules are dilated.
The disease is asymptomatic in most cases, and is often an accidental finding during USG. Symptoms include hip or abdominal pain, hematuria, urinary tract infections, nephrolithiasis, hypertension, liver cysts, mitral valve prolapse, intracranial aneurysm, or progressive deterioration in renal function. The above symptoms do not appear until adolescence or middle age.
The diagnosis should be considered in patients who have positive family history and have even mild proteinuria or micro-/ macrohematuria. The diagnosis can be made using USG, excretory urography, or CT. ADPKD is also diagnosed when a detective gene is found.
There is no causal therapy. Chronic renal insufficiency, hypertension, and urinary tract infections are treated if present as well. Progressive deterioration of renal function in a patient with ADPKD is an indication for transplantation.
Autosomal recessive polycystic kidney disease[edit | edit source]
Autosomal recessive polycystic kidney disease (ARPKD) is a juvenile form of PKD and is much rarer (incidence 1:20 000–40 000) and is caused by a gene mutation in chromosome 6.
It is manifested by the rapid development of renal insufficiency in the postpartum period. In children with this disease, pulmonary hypoplasia, oligohydramnios, or liver fibrosis occur at an early age with the development of portal hypertension.
The disease is diagnosed by ultrasound imaging of the kidney or prenatally using specific prenatal diagnostic techniques. It is possible to do so from the 24th week of pregnancy.
Diagnosis is important for appropriate therapy during the postpartum period. Later on, CKD or liver failure therapy is initiated.
Gallery[edit | edit source]
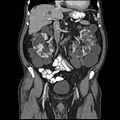
CT of polycystic kidney disease

MRI image of polycystic kidney disease
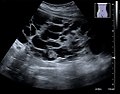
USG imaging of a polycystic kidney



Summary video[edit | edit source]
References[edit | edit source]
Literature[edit | edit source]
- DÍTĚ, P.. Vnitřní lékařství. 2. edition. Praha : Galén, 2007. ISBN 978-80-7262-496-6.
