Wilms' tumor


| File:Nefroblastom, 6ti leta pacientka.png |
Wilms tumor or nephroblastoma (WT, OMIM: 194070) is a relatively common solid tumor of childhood. Specifically, it is a kidney cancer (unilateral or bilateral) characterized by clusters of embryonic nephrogenic blastoma cells. They make up about 5-6% of childhood tumors and are the sixth most common tumor in children. It typically affects preschool children, with a maximum incidence between the 2nd and 3rd year of life. It affects both kidneys in about 7 % of cases.[1][2]
It is very often associated with congenital defects. These include urogenital tract defects, aniridia, pseudohermaphroditism, macroglossia.
Etiopathogenesis[edit | edit source]
Only about 1% of cases have a hereditary cause, the vast majority of cases arise sporadically. Hereditary form is mainly associated with mutated WT1 gene on 11th. chromosome (11p13). Rarely, mutations in other genes may be involved (WT2–WT5). WT1 tumor-supressor gene that codes a transcription factor (type zinc finger) is involved in the differentiation of the urogenital tract. Hereditary syndrome sometimes occurs in association with sporadic aniridia (lack of iris), sometimes also in association with neurofibromatosis type 1, mutations of BRCA1 gene či Bloom syndrome. Congenital syndromes that have a high risk of developing WT include Beckwith-Wiedemann - hemihypertrophy of the limbs and Drash's. Children with these syndromes must be monitored by an oncologist for at least the first 6 years of life.[1][2]
Clinical picture[edit | edit source]
- The most common manifestation is painless resistance in the abdomen; parents may sometimes notice an increase in tummy tuck ("tight pants");
- non-specific problems - anorexia, constipation, vomiting, fever;
- approximately in 20 % of children the first manifestation is hematuria[2] or abdominal pain.
Metastases[edit | edit source]
- hematogenous spread – mainly the lungs, then the liver, the brain. Rarely to the bone;
- Lymphogenous spread – to regional lymph nodes (hilar, paraaortic).
Diagnostics[edit | edit source]
- Ultrasound examination of the abdomen;
- CT examination of the abdomen;
- X-ray and CT of lungs to rule out metastases;
- the definitive diagnosis is histological (surgical solution usually follows chemotherapy)[2];
- the hereditary form can be confirmed by targeted molecular genetic testing (available in the Czech Republic).
Therapy[edit | edit source]
- Usually neoadjuvant chemotherapy – 4 weeks;
- surgical removal of the entire kidney with tumor and regional nodes;
- chemotherapy – actinomycin D, vincristine, cyclophosphamide..;
- event. radiotherapy[2];
- high-dose chemotherapy followed by bone marrow transplantation – treatment of recurrences after reaching 2nd or 3rd remission.
Special approach required in treatment of bilateral tumors:
- nephrectomy of a more affected kidney and partial nephrectomy on the other hand are indicated;
- bilateral nephrectomy with kidney transplantation.
Prognosis[edit | edit source]
- It can be cured in 90 % of children, if the tumor is inside the kidney;
- if the tumor is bilateral or spread beyond the kidney, 60 % of children survives.[2]

Wilms' tumor (nefroblastoma)

CT scan – Wilms' tumor
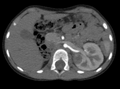
CT scan - nephroblastoma in a 4-year-old patient with a solitary double kidney



Summary video[edit | edit source]
Links[edit | edit source]
Related articles[edit | edit source]
Source[edit | edit source]
- ŠÍPEK, Antonín. Geneticky podmíněná nádorová onemocnění [online]. The last revision 8. 6. 2007, [cit. 4. 2. 2010]. <http://www.genetika-biologie.cz/hereditarni-nadorove-syndromy>.
Reference[edit | edit source]
- ↑ a b KLEIBL, Zdeněk – NOVOTNÝ, Jan. Hereditární nádorové syndromy. 1. edition. Praha : Triton, 2003. 31 pp. ISBN 80-7254-357-1.
- ↑ a b c d e f Klinika dětské onkologie FN Brno. Kidney tumors - nefroblastoma (Wilms tumor) [online]. [cit. 2011-01-02]. <https://www.fnbrno.cz/detska-nemocnice/klinika-detske-onkologie/informace-pro-pacienty/t2698>.

{kind=link}