Congenital defects of the nervous system: Difference between revisions


Vlckovaanna2 (talk | contribs) |
Vlckovaanna2 (talk | contribs) |
||
| Line 110: | Line 110: | ||
<noinclude> | <noinclude> | ||
==[[ | ==[[Meningocele]]== | ||
[[File:Typesofspinabifida.jpg|thumb|right|400px|Types of spinal disorders]] | [[File:Typesofspinabifida.jpg|thumb|right|400px|Types of spinal disorders]] | ||
'''Meningocele''' ''(spina bifida cystica)'' is a split of the vertebral arch, from which emerges a sac made up of soft [[brain coverings|spinal cords]] (the dura mater usually ends at the neck of the sac). The content is [[cerebrospinal fluid]], 1/3 of children are neurologically affected. | '''Meningocele''' ''(spina bifida cystica)'' is a split of the vertebral arch, from which emerges a sac made up of soft [[brain coverings|spinal cords]] (the dura mater usually ends at the neck of the sac). The content is [[cerebrospinal fluid]], 1/3 of children are neurologically affected. | ||
| Line 116: | Line 116: | ||
'''Menigomyelocele'' ''(spina bifida aperta)'' is a variant of meningocele that occurs a little more often and is less favorable. In addition to the packages, there is also [[spinal cord]] in the hernial sac. | '''Menigomyelocele'' ''(spina bifida aperta)'' is a variant of meningocele that occurs a little more often and is less favorable. In addition to the packages, there is also [[spinal cord]] in the hernial sac. | ||
====Therapy==== | ====Therapy==== | ||
On admission, place the child on the stomach and exclude pressure on the meningomyelocele. In case of perforation, apply [[Antibiotics|ATB]] and cover the bearing with a mule. We operate within 24 hours. In complete motor paraplegia and sphincter lesions, this cannot be improved by any surgery, but surgery is in order to prevent infection. | On admission, place the child on the stomach and exclude pressure on the meningomyelocele. In case of perforation, apply [[Antibiotics|ATB]] and cover the bearing with a mule. We operate within 24 hours. In complete motor paraplegia and sphincter lesions, this cannot be improved by any surgery, but surgery is in order to prevent infection. | ||
[[Category:Neurosurgery]] | [[Category:Neurosurgery]] | ||
[[Category:Neurology]] | [[Category:Neurology]] | ||
| Line 165: | Line 122: | ||
[[Category:Pediatrics]] | [[Category:Pediatrics]] | ||
[[Category:Neonatology]] | [[Category:Neonatology]] | ||
<noinclude> | |||
< | |||
== [[Neurofibromatóza]]== | == [[Neurofibromatóza]]== | ||
Revision as of 15:17, 16 December 2022
Arachnoid cyst
An arachnoid cyst is a well-defined cystic, congenital collection of cerebrospinal fluid (it arises on the basis of doubling of the arachnoid layer). It is often an accidental finding on a CT scan or MRI scan. It is important for a differentia diagnosis of (tumors).[1] [2]
Symptoms and auxiliary tests
In the CT image (resp MRI, possibly with the intrathecal application of contrast) arachnoid cyst has a density, resp. cerebrospinal fluid intensity (hypodense). Most common locations:
- in the middle pit
- on convexity
- in the posterior cranial pit (cisterna magna)
- in the cerebellopontine angle
- at the chiasm level
- in the chamber
Expansive behaviour depends on its communication with the surrounding cerebrospinal fluid spaces. The problem occurs if the communication has a valve character (intracranial hypertension syndrome) occurs.
Arachnoid cysts can be asymptomatic, otherwise, they often cause symptoms in early childhood (increased intracranial pressure, epileptic seizures, gradually developing focal neurological symptoms, sudden worsening of the condition during cyst bleeding.[1][2]
- CT of arachnoid cysts of the temporal lobe

Sagittal plane

Frontal plane
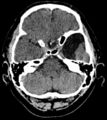
Transverse plane



Treatment
An accidental finding and an asymptomatic cyst are not treated or followed up. For expansive behaviour and clinical manifestations, surgery is chosen (shunt surrounding cerebrospinal fluid, ie microsurgical / endoscopic fenestration of the cyst into the normal cerebrospinal fluid). A shunt formed between the arachnoid cyst (most often in the case of the sulcus lateralis cyst) and the peritoneal cavity, the so-called cystoperitoneal shunt, is also possible. Marsupialization (removal of the cyst with the capsule) is rarely performed.[1][2]

Chiari malformation
Chiari malformation (Arnold-Chiari malformation) is a congenital CNS anomaly. It is a dystopia of the cerebellum and medulla oblongata into the spinal canal, which is clinically manifested by hydrocephalus. We distinguish four types of rhombencephalon abnormalities (cerebellum, pons, oblongata):
- Type 1 - herniation of the cerebellar tonsil under the foramen magnum, IV. the chamber is stored normally
- Type 2 - usually the co-presence of myelomeningocele
- Type 3 - severe dislocation of structures in the posterior pit, often associated with suboccipital encephalomeningocele; usually incompatible with life
- Type 4 - cerebellar hypoplasia without herniation.

Clinical picture
Clinically, the defect is manifested mainly by headache, weakened grip and spasticity of DK.
Diagnosis
Native X-ray, MRI.
Therapy
The main problem is hydrocephalus – decompression of the craniospinal junction, short-circuit drainage operations.
Craniostenosis

Craniostenosis is caused by premature adhesion of sutures. The earlier it occurs, the more severe its effects. The shape of the head depends on the order in which the sutures close - as a suture grows, the skull stops growing perpendicular to the suture.
Normal development of the skull and sutures
- at birth, the skull has one lamina, and the diploe is formed at 4 years of age
- large fontanelle is 4x2.5 cm at birth, closes at 1.5-2.5 years
- the small one closes at 2-3 months
- the growth of the sutures is prevented by the proliferation of fibroblasts along the suture line
- CI - cephalic index - percentage ratio of head width to length - 60-70% in children, 70-90% in adults
- lengthening of the head decreases CI
Dividing
- primary - due to a developmental disorder of unclear etiology;
- secondary - in microcephaly and after drainage hydrocephalus.
Diagnosis
- clinically - palpation of fontanelles, X-ray, CT
- at high pressure, gyral impressions are seen on the skull bones - the so-called skull of wrought silver
- scintigraphy - open suture receives Tc, closed not
Types
- scaphocephaly - sagittal suture fusion, the head is narrow, resembles a ship's keel
- brachycephaly - affects the coronal suture - flat forehead, greater distance between orbits, etc.
Therapy
- many surgical methods, indications are intracranial hypertension or cosmetic
- early surgery can ensure normal psychomotor development
Meningocele

Meningocele (spina bifida cystica) is a split of the vertebral arch, from which emerges a sac made up of soft spinal cords (the dura mater usually ends at the neck of the sac). The content is cerebrospinal fluid, 1/3 of children are neurologically affected.
'Menigomyelocele (spina bifida aperta) is a variant of meningocele that occurs a little more often and is less favorable. In addition to the packages, there is also spinal cord in the hernial sac.
Therapy
On admission, place the child on the stomach and exclude pressure on the meningomyelocele. In case of perforation, apply ATB and cover the bearing with a mule. We operate within 24 hours. In complete motor paraplegia and sphincter lesions, this cannot be improved by any surgery, but surgery is in order to prevent infection.
Neurofibromatóza
Template:Infobox - genetická choroba
Neurofibromatóza je relativně časté AD dědičné onemocnění (1:2 500–4 000 novorozenců) vycházející z buněk odvozených z neurální lišty. Projevuje se abnormálním růstem podpůrných buněk CNS a PNS (Schwannovy bb. aj.) s výraznou predispozicí k vzniku benigních i maligních nádorů.[3] Onemocnění patří mezi hereditární nádorové syndromy, vyskytuje se ve dvou formách. Familiární formy těchto syndromů vznikají jako následek vrozené mutace tumor-supresorových genů. Určité procento těchto syndromů vzniká jako následek nových mutací.[4] Molekulárně-genetická analýza je dostupná pro definitivní potvrzení této diagnózy.
náhled|vpravo|Časné známky neurofibromatózy typu I: drobné neurofibromy a skvrny barvy bílé kávy (café-au-lait) náhled|Neurofibrom 7 cm náhled|Lischovy uzlíky v duhovce
Formy
Existují dvě základní formy tohoto onemocnění, které se liší jak v příčině (mutace různých genů), tak i v následcích (odlišný klinický obraz).
Neurofibromatóza – typ 1
Neurofibromatóza – typ 1 (NF-1, nazývaná též morbus von Recklinghausen či periferní typ neurofibromatózy) je podmíněna mutací NF1 genu na 17. chromosomu (17q11.2).[5] Jde o tumor-supresorový gen, jehož produkt (neurofibromin) je součástí intracelulární signální kaskády spojené s RAS-kinasou.
![]()
Mezi klinické projevy této formy patří:
- Tzv. „café-au-lait spots“ (skvrny barvy „bílé kávy“, v 90 % se objeví do 5 let věku)[4]
- Neurofibromy (mnohočetné tumorózní uzlíky; kutánní, subkutánní a plexiformní; hlavně v axilách a tříslech)
- Lischovy uzlíky (hamartomy duhovky)[4][5]
- Zvýšené riziko vzniku různých nádorových onemocnění: gliomy CNS (gliomy optiku), neurofibrosarkom, rhabdomyosarkom, feochromocytom, leukemie apod.[4]
- Postižení muskuloskeletáního systému (subperiostální neurofibromy – působící hypertrofii kosti, její prořídnutí a pathologické fraktury, skolióza, vrozená dysplázie tibie)[3]
- Postižení intelektu, epilepsie nebo stenózy arteria renalis [4][5]
Neurofibromatóza – typ 2
Neurofibromatóza – typ 2 (NF-2, nazývaná též MISME syndrom či centrální typ neurofibromatózy) je podmíněna mutací NF2 genu na 22. chromosomu (22q12.2)[5]. Jde rovněž o tumor-supresorový gen, jehož produkt (neurofibromin 2, též nazývaný merlin či schwannomin) ovlivňuje mezibuněčné kontakty. Centrální neurofibromatóza je obecně vzácnější než periferní typ, celkově je ovšem spojena s vyšší morbiditou i mortalitou postižených jedinců[4]. Okolo poloviny případů NF-2 je způsobeno novou mutací[5].
Mezi klinické projevy této formy patří:
- Nádory CNS: meningeomy, astrocytomy, ependymomy, schwanomy míšních kořenů, hamartomy sítnice (syndrom MISME = Multiple Inherited Schwannomas, Meningiomas, and Ependymomas)
- Typický je zejména bilaterální vestibulární schwannom
- Také u této formy nacházíme skvrny „café-au-lait“, ne však Lischovy uzlíky[4]
náhled|vpravo|180px|RTG neurofibromatóza – patrné neurofibromy
Terapie
- Kauzální terapie neexistuje.
- Vhodná je dispenzarizace pacientů s prokázanou diagnózou neurofibromatózy.
- Chirurgické zákroky indikovány při útlaku nervu / obstrukci u GIT formy, případně z kosmetického hlediska[3].
- Neurochirurgické zákroky při postižení CNS; možné využití stereotaktické neurochirurgie (Leksellův gama nůž).
Odkazy
Související články
- Hereditární nádorové syndromy
- Tumor-supresorové geny
- Vrozené mnohočetné exostózy
- Enchondromatóza (Ollierova choroba)
- Fibrózní kostní dysplázie (Jaffé-Lichtensteinova nemoc)
- Osteogenesis imperfecta (osteopsatyrhosis, fragilitas ossium)
- Morbus Albers-Schönberg (mramorovitost kostí, osteoskleróza, osteopetróza)
- Osteopoikilóza (osteopoikilie)
Externí odkazy
- Neurofibromatosis – Type 1 (eMedicine)
- Neurofibromatosis – Type 2 (eMedicine)
- The Children's Tumor Foundation
Reference
- ↑ Jump up to: a b c SEIDL, Zdeněk – OBENBERGER, Jiří. Neurologie pro studium i praxi. 1. edition. Praha : Grada Publishing, 2004. ISBN 80-247-0623-7.
- ↑ Jump up to: a b c SAMEŠ, M, et al. Neurochirurgie. 1. edition. Praha : Jessenius Maxdorf, 2005. ISBN 80-7345-072-0.
- ↑ Jump up to: a b c
- ↑ Jump up to: a b c d e f g
- ↑ Jump up to: a b c d e
Template:Navbox - monogenně dědičné choroby
Kategorie:Ortopedie
Kategorie:Neurologie
Kategorie:Neurochirurgie
Kategorie:Genetika
Kategorie:Patologie
Dandyova-Walkerova malformace
__ Dandyova-Walkerova malformace
Template:Infobox - genetická choroba Dandy-Walker syndrom je vzácné onemocnění, které postihuje vývoj mozku. Projevuje se triádou příznaků
- kompletní nebo parciální agenesí vermis mozečku
- cystická dilatace čtvrté komory mozkové
- zvětšení zadní jámy lební[1]
Zvětšení zádní jámy může být spojeno s membranózní atrezií otvorů čtvrté mozkové komory, čímž dochází k patologickému hromadění mozkomíšního moku v mozkových komorách. Tyto abnormality mají nejčastěji za následek problémy s pohybem, koordinací ale i s intelektem. Může se taky vyskytnout psychiatrické onemocnění[2]. Postižení se mohou dožívat až druhého decenia.
Malformace
náhled|200px|Ageneze mozečku Většinou se u jedinců s Dandy-Walker syndromem objevují příznaky abnormálního vývoje mozku v průběhu prvního roku života. Klinicky se u většiny dětí projevuje hromaděním mozkomíšního moku v mozku – hydrocefalem, který může způsobit makrocefalii. Často je přítomno mentální postižení[3] , ačkoli někteří jedinci mají i normální intelekt. Děti mají často opožděný vývoj, zejména v oblasti motoriky (plazení, chůze, koordinace pohybů). Může se objevovat ztuhlost svalů a ochrnutí – spastická paraplegie. U starších dětí se objevují příznaky zvýšeného nitrolebního tlaku jako je podrážděnost, zvracení, křeče a trhavé pohyby očima. Méně často jsou přítomny jiné mozkové malformace jako je ageneze corpus callosum spojujícím pravou a levou hemisféru, okcipitální encefalokéla nebo abnormální štěrbiny v mozku – schizencephaly a poruchy gyrifikace. Tyto mozkové vady jsou spojeny s více či méně závažnými příznaky a symptomy. U Dandy–Walker syndromu se mohou vyskytovat i srdeční vady, malformace urogenitálního traktu, končetin a obličeje. Mírnější formou onemocnění je Dandy–Walker varianta zahrnující hypoplasii vermis mozečku bez agenese, mírné či žádné zvětšení zadní jámy lební a čtvrté komory, nebývá hydrocefalus.
Genetika
U jedinců postižených Dandy-Walker syndromem se objevují mutace v určitých genech, ale tyto mutace představují pouze malý počet všech zjištěných případů. Dandy-Walker syndrom je také spojován s chromozomálními abnormalitami u většiny chromozomů. Nejčastěji se vyskytuje u lidí s trizomií 18, ale může se také vyskytovat trizomie 13, 21 nebo 9. Dandy-Walker syndrom byl také zaznamenán u plodů s triploidií, což je fatální stav, při němž mají jedinci navíc kompletní sadu chromozomů v každé buňce. Dandy-Walker syndrom může být také způsoben mutacemi specifických genů (FOXC1 obsahuje pokyny pro přípravu proteinu, který reguluje aktivitu dalších genů, ZIC1 a ZIC4 působí jako transkripční aktivátory a podílejí se na organogenesi CNS). Mozkové malformace spojené s Dandy-Walker syndromem se mohou vyskytovat izolovaně, nejsou spojeny s genetickým onemocněním a příčina vzniku bývá často neznámá.
Vliv prostředí
Dandy-Walker syndrom může být způsoben i faktory životního prostředí, které působí na plod před jeho narozením. Například expozicí plodu zarděnkami, toxoplasmosou nebo vlivem látek, které způsobují poškození plodu (teratogeny). Častější výskyt plodů s Dandy-Walker syndromem se objevuje i u plodů matek, které trpí diabetem.
Diagnostika a výskyt
Prenatální diagnostika je možná ultrazvukovým vyšetřením a provedením amniocentesy k vyšetření karyotypu plodu. Odhaduje se, že se Dandy-Walker syndrom vyskytuje u 1:25000 až 1: 30000 novorozenců. Výskyt případů Dandy-Walker syndromu je málo častý a nemá jasný vzor dědičnosti. Pouze přímí příbuzní lidí s Dandy-Walker syndromem mají zvýšené riziko vzniku onemocnění ve srovnání s běžnou populací.
Terapie
Terapie spočívá v odstranění obstrukce, hydrocefalus léčíme zavedením shuntu.
Odkazy
Související články
Externí odkazy
Použitá literatura
Reference
Kategorie:Neurologie
Kategorie:Neurochirurgie
Kategorie:Pediatrie
Dětská mozková obrna
Dětská mozková obrna (DMO, angl. cerebral palsy) je trvalá neprogredující porucha hybnosti provázená abnormálním svalovým napětím a abnormální posturou. Jedná se o nenakažlivé, nedědičné onemocnění, které vzniká na podkladě jednorázového poškození mozkové tkáně (nejčastěji hypoxií). Je charakteristické poruchou vývoje motorických oblastí mozku nebo jejich jiným poškozením v raném stádiu vývoje. DMO je následkem prenatální, postnatální nebo raně postnatálního poškození vyvíjejícího se mozku.
K poruše hybnosti se mnohdy připojuje epilepsie, poruchy citlivosti, smyslů (např. zrakové postižení) a vnímání, poruchy učení, kognice, komunikace, chování nebo mentální retardace. DMO je neurovývojovou poruchou, jejíž projevy se v průběhu vývoje většinou mění.
DMO patří do skupiny vývojových onemocnění, protože vzniká na základě širokého spektra abnormalit vyvíjejícího se CNS. Různé etiologie působící na různá vývojová stadia mohou vést ke stejnému klinickému obrazu a naopak obdobná etiologie může vyvolat odlišné následky.
Léčba DMO je komplexní, multioborová a dlouhodobá. Jejím cílem není vyléčení nebo dosažení normálního stavu, ale zvětšení funkčnosti, zlepšení schopností a udržování zdraví ve smyslu lokomoce, kognitivního vývoje, sociální integrace a nezávislosti. Úspěch terapie závisí na její včasnosti a intenzitě.
![]()
Klasifikace
- podle anatomické topografie postižení:
- mono-, hemi-, di-, kvadruparetická forma;
- podle patofyziologického typu hybné poruchy:
- spastické formy a nespastické formy: s dyskinezami, dystoniemi, hypotonií, ataxií.[1]
Epidemiologie
Incidence DMO je asi 2:1000 živě narozených dětí. Riziko roste nepřímo úměrně s gestačním stářím při porodu.[2]
Etiologie

Hybnou poruchu při dětské mozkové obrně způsobuje postižení supraspinálních hybných center, kortikospinálních traktů, segmentálních spinálních okruhů a muskuloskeletárního systému.[1]
Může vzniknout během jednoho ze čtyř následujících období:
- V těhotenství (prenatální etiologie) – intrauterinní infekce, léky nebo placentární dysfunkce;
- Při porodu (perinatální etiologie) – hlavně u vícečetných těhotenství, hypoxie, hemoragie, hypoglykémie, meningitida;
- V prvních měsících života dítěte (postnatální etiologie) – trauma, encefalopatie, encefalitida;
- Popř. velmi nízká porodní hmotnost.
V prvním a druhém trimestru vznikají poruchy vývoje CNS. Na začátku třetího trimestru to je periventrikulární leukomalácie (PVL) a intraventrikulární hemoragie (IVH). Ke konci třetího trimestru vznikají léze kortikální, subkortikální a léze hluboké mozkové šedi.[1]
- Bilaterální spastické a dyskinetické formy – hypoxicko-ischemická léze mozku.
- Diparéza – periventrikulární leukomalacie u předčasně narozených dětí.
- Spastická hemiparéza zralých novorozenců – infarkt a. cerebri media, periventrikulární glióza.
- Spastická hemiparéza nezralých novorozenců – periventrikulární porencefalie v důsledku intraventrikulárního krvácení.
- Dyskinetická forma – hypoxicko-ischemické poškození v oblasti thalamu a bazálních ganglií (děti po 32. týdnu těhotenství).
- Atetoidní forma – neonatální hyperbilirubinémie s jádrovým ikterem (velmi vzácná).
- Ataktická forma – většinou neznámá příčina, ev. strukturální porucha mozečku.[2]
Klinické příznaky

- forma: spastická, dyskinetická nebo ataktická;
- distribuce: bilaterální – diparéza, kvarduparéza, unilaterální – hemiparéza;
- míra postižení: škála GMFCS I – V (Gross Motor Function Classification System) – funkční test pro hodnocení míry a následných změn v hrubé motorice pomocí standardních volních pohybů (volní hybnost, chůze a sed); MACS (The Manual Ability Classification System) – test manuálních schopností;
- komorbidity: epilepsie, mentální retardace, smyslové poruchy.[1]
- Spasticita: abnormálně zvýšený svalový tonus, zvýšené svalové reflexy, pozitivní pyramidové jevy, abnormální pohyby a držení (pes equinus, vnitřní rotace a addukce v kyčlích, pronace a flexe předloktí), vznik kontraktur.
- Dystonie: abnormální trvalé svalové kontraktury, které vedou k abnormálnímu dystonnímu držení a abnormálním pohybům (flexe, pronace zápěstí s nataženými prsty nebo torzí trupu).
- Atetóza: generalizované, nekoordinované, přehnané, mimovolní hyperkinetické pohyby, normální nebo snížený svalový tonus.
- Ataxie: dysmetrie nebo intenční tremor horních končetin, ataxie chůze a stoje – postižení dolních končetin a trupu (stoj a chůze o široké bazi, kolíbavá chůze).[2]
Spastické formy DMO
Diparetická forma (někdy označována jako paraparetická)
- Nejčastější (1/3 postižených).
- Symetrické postižení obou dolních končetin (slabší, méně vyvinuté).
- Nápadný nepoměr mezi vzrůstem trupu a dolních končetin.
- Svalová hypertonie, zkraty svalů – vadné držení dolních končetin i pánve.
- Může být lehká až střední mentální retardace.
Hemiparetická forma
- Druhá nejčastější.
- Postižené končetiny slabší a zpravidla kratší ve srovnání s druhostrannými.
- Skoro vždy více postižena horní končetina.
- Typické postižení charakterizováno paží přitaženou k trupu, je pokrčena až úplně ohnutá v lokti, předloktí otočeno hřbetní stranou vzhůru, ruka ohnuta směrem do dlaně a uchýlena směrem k malíkové straně.
- Častý je výskyt epilepsie, případně může být i senzitivní deficit a mentální retardace.
Kvadruparetická forma
- Těžší forma diparetické formy.
- Obrnou postiženy všechny 4 končetiny.
- Přítomna je i epilepsie a mentální retardace či poruchy učení.
Dyskineticko-ataktické formy DMO
- Cca 10–15 % případů DMO.
- Epilepsie je u těchto forem vzácná.
- Převládá-li choreo-atetóza, bývá normální inteligence, ale slovní vyjadřování ztěžuje těžká dysartrie.
- Převládá-li dystonie nebo ataxie, častěji je přítomna i mentální retardace.
Onemocnění je často kombinací více forem DMO.
Diagnostika
- DMO je klinická diagnóza; definitivně stanovena je až po 3. roce věku (při nezralosti CNS jsou projevy nespecifické).
- Vyšetření k objasnění etiologie:
- UZ mozku, MR mozku;
- koagulace (protein C a S, homocystein, APC rezistence) – při prokázaném infarktu;
- EEG; vyšetření zraku a sluchu;
- genetické vyšetření – u lissencefalie.[2]
Diferenciální diagnostika
Pro DMO svědčí hlavně stabilní klinický nález bez progrese postižení. Pro stanovení rozsahu je důležité zobrazovací vyšetření (CT, MRI). Progrese postižení ukazuje spíše na neurodegenerativní či neurometabolické postižení. Pomalu rostoucí tumor CNS.
Terapie
DMO je choroba, která se nedá vyléčit. Dá se však pomocí léčby dosáhnout zlepšení podmínek a životních možností dítěte, což většinou příznivě ovlivňuje kvalitu jeho života. Lékařské pokroky posledních let a hlavně pokroky v léčbě postižených s DMO vedly k tomu, že dnes již mnozí z těch, kteří byli včas a správně léčeni, mohou vést téměř normální život. Ani dnes ještě neexistuje nějaká standardní léčba, která by byla u všech pacientů dostatečně účinná. Lékař, který léčí pacienta s DMO, je víceméně závislý na řadě specializovaných odborníků, s jejichž pomocí nejprve správně určí a stanoví individuální poruchy a pak jim přizpůsobí celý terapeutický program. V současné době probíhá také několik klinických studií na aplikaci kmenových buněk z pupečníkové krve.
Léčebný plán může zahrnovat:
- Rehabilitaci – zlepšení kontroly lokomoce i posturálního držení pomocí stimulačních a facilitačních technik (např. Vojtovou metodou), nejprve odborníky, posléze instruovanými rodiči.
- Antiepileptika.
- Myorelaxans u spastických forem.
- Dlahy pro svalovou nerovnováhu.
- Operační léčbu – ortopedické korekce (mezi 6. a 10. rokem), například u diparetické formy selektivní dorzální rizotomie (částečné přetětí zadních míšních kořenů ve výši L2).
- Protetické pomůcky.
- Aplikaci botulotoxinu.
- Speciální péči a výchovu od předškolního věku.
Odkazy
Související články
Externí odkazy
Použitá literatura
- ↑ Jump up to: a b c d
- ↑ Jump up to: a b c d
Kategorie:Neurologie Kategorie:Pediatrie
Odkazy
Související články
Zdroj
Reference
Kategorie:Neurologie
Kategorie:Embryologie
Kategorie:Genetika

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}